1Departmentof Pediatric Endocrinology, CHU Hassan II Fez, Morocco.
2Department of Endocrinology & Diabetology, CHU Hassan II Fez, Morocco.
3Deparment of Mother Child Radiology, CHU Hassan II Fez, Morocco.
3Department of Faculty Medicine and Pharmacy, CHU Hassan II of Fez, Morocco.
Zineb Essolaymany
Email: slimani.zinebs@gmail.com
Received : Dec 17, 2024 Accepted : Jan 17, 2025 Published : Jan 24, 2025 Archived : www.meddiscoveries.org
Pituitary stalk interruption syndrome, an uncommon cause of growth hormone deficiency and hypopituitarism, is often revealed during the neonatal period and in childhood. The association with Kallmann syndrome (a rare genetic disease, associating hypogonadotropic hypogonadism and anosmia or hyposomnia secondary to abnormal and altered migration of GnRH neurons) is very rare.
Birth We report the case of a 13-year-old child admitted for stature-weight delay with no particular neonatal history. Clinical examination revealed severe growth retardation with micropenis. The hypophysiogram showed complete hypopituitarism with diabetes insipidus. Hormone replacement therapy was administered to our patient. The association of the two pathologies, pituitary stalk interruption syndrome and Kallmann’s syndrome (agenesis of the olfactory bulbs), suggests the possibility of a common genetics and pathophysiology, hence the need for further research in this field.
Keywords: Pituitary Stalk Interruption Syndrome (PSIS); aplasia/hypoplasia of the Olfactory Bulbs (OB); Kallmann syndrome.
Kallmann Syndrome (KS) is characterized by Congenital Hypogonadotropic Hypogonadism (CHH) and anosmia/hyposmia associated with aplasia/hypoplasia of the Olfactory Bulbs (OB) [1]. In KS, gonadotropin deficiency is isolated, statural growth is preserved and other pituitary functions, notably Growth Hormone (GH) secretion, are normal, as is hypothalamic-pituitary morphology [2].
Pituitary Stalk Interruption Syndrome (PSIS) is a congenital pituitary anatomical defect. It is characterized by the triad of a thin or interrupted pituitary stalk, an absent or ectopic posterior pituitary gland, and a hypoplastic or aplastic anterior pituitary gland. This anatomical anomaly may be associated with extra-pituitary morphological abnormalities and variable endocrine disorders, ranging from isolated GH deficiency to Combined Pituitary Hormone Deficiency (CPHD) [3].
To date, neither OB abnormalities in PSIS patients nor morphological abnormalities of the pituitary stalk in SK patients have been proven. The different pathophysiologies of KS and PSIS, which involve altered GnRH neuron and olfactory system development in KS [1,2], and pituitary organogenesis in PSIS, have been proposed as the causes of phenotypic differences between the two disorders [3,4]. However, recent work has challenged these notions, suggesting that sometimes SK and PSIS may share a common genetic origin and pathophysiology [4]. we demonstrate here a sporadic case of KS associated with PSIS that reinforces this hypothesis.
A 13-year-old patient was followed up for severe stature-weight delay (>-3DS). His mother noted that the patient was small in stature compared with children of the same age. But there was no detailed data on the patient’s height and weight over the course of his development. Moreover, the patient also had a severe micropenis with hypoplastic testes, pubertal stage G1P1 according to Tanner classification.
This patient was the first child of a first-degree consanguineous couple; he was born full term via caesarean delivery for fetopelvic disproportion. His birth weight and length were normal, and he had no significant medical or family history of short stature, and his younger brother was growing normally. On questioning, the patient stated that he also had anosmia from birth, which had never been explored.
Hormonal tests indicated pituitary insufficiency: IGF1 levels were very low (28 ng/ml), cortisol levels were very low (1.3 ug/dl), TSH was normal with a low LT4 (0.52), and the GH stimulation test revealed growth hormone deficiency. Exploration of the gonadotropic axis with testosterone, LH, and FSH is planned at puberty.
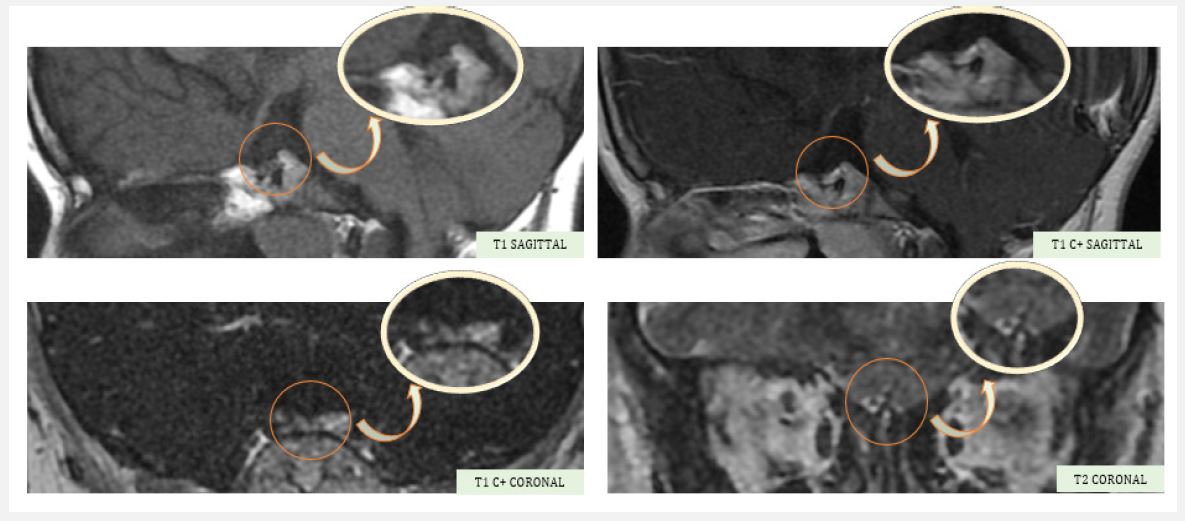
A hypothalamo-pituitary MRI was ordered, revealing an anterior pituitary gland in place, a decrease in height measured at 2 mm, a homogeneous signal before and after contrast, and a lack of clear visualization of the pituitary stalk and the posterior pituitary gland, associated with very severe hypoplasia of the olfactory bulbs, suggestive of Kallmann syndrome.
An analysis of the postpituitary gland revealed diabetes insipidus, and the patient was put on minirin, and hormone replacement with l-thyroxine, hydrocortisone, and growth hormone was prescribed. For his micropenis, he was given a micropenis cure with good clinical and biological evolution.
Congenital Hypogonadotropic Hypogonadism (CHH) accompanied by anosmia or hyposmia is the phenotypic feature of the developmental disorder known as Kallmann Syndrome (KS) [5].
KS occurs when the migration of embryonic gonadotropin-releasing hormone neurons from the nasal placode to the hypoathalamus is disrupted.
The disease affects 1 in 8,000 boys, and is five times more common in men than in women. Different types of inheritance have been explained, including X-linked recessive, autosomal dominant and autosomal recessive, but SK is often sporadic.
The clinical expression of the same genetic mutation can vary considerably, from simple pubertal delay and normosmia to complete anosmia with hypogonadotropic hypogonadism [6,7].
The molecular basis of KS has undergone considerable progress over the last 30 years, which partly explains the variety of phenotypic abnormalities.
KAL1 mutations contribute to the formation of X-linked SK and fibroblast growth factor. Mutations in the type 1 Receptor (FGFR1) cause an autosomal dominant form of KS. Only 20 to 25% of KS cases are caused by mutations in these two genes [2]. Several other mutations with phenotypic differences have been described in the literature (PROK2 [11,14,15]; CHD7 [16]; SEMA3 [17]; SOX10 [11].
Clinically, micropenis, loss of voice change, permanent hair loss and infertility are common symptoms of KS. Anosmia is most often diagnosed.
With KAL1 mutation, 30% of cases have renal agenesis and 75% of cases have mirror movements [5]. This mutation is characterized by an arched palate [5].
A distinctive feature of KS is short stature. Kallmann syndrome was first identified as one of the causes of short stature by Subramanian et al [8]. Since then, few cases of small-sized SK have been reported, but the olfactory bulbs were usually normal [9].
Paraclinically, MRI is the modality of choice for evaluating the olfactory bulbs. It is advisable to use a coronal scan with a large matrix size and reduced intersection space to optimally visualize the olfactory bulbs [10].
Koenigkam-Santos et al. found that the most common signs in KS patients were aplasia of the olfactory bulb and sulcus. Lack of GnRH stimulation of the anterior pituitary lobe may be related to pituitary hypoplasia [11].
Initial treatment for adolescents and young men with SK is to increase penis size, masculinization of the voice, development of muscle mass and hair growth. In addition, treatment helps to increase libido and change sexual behavior [12].
Correction of delayed bone maturity and prevention of osteoporosis are two other therapeutic targets. Testosterone therapy is more effective than pulsatile GnRH or combined gonadotropin therapy. One of the most important parts of care is counseling the patient and family regarding the patient’s clinical condition and long-term management.
The congenital malformation known as pituitary stalk interruption syndrome was first described in 1987 by Fujisawa et al. Mutations in genes encoding transcription factors involved in anteropituitary ontogeny have been reported, the most frequently implicated being PROP1, POUIF1, HESX1, LHX3, and LHX4. Its etiopathogenesis remains unknown, and it manifests clinically as a single or multiple pituitary deficiency [13].
The progressive nature of the disease requires reassessment of the various pituitary axes, especially as the deficits become intense and multiple over time [14].
This syndrome is defined by morphological abnormalities highlighted by magnetic resonance, namely a small or non-visible pituitary stalk, anteropituitary hypoplasia, and ectopic posthypophysis [15].
Our case report is the second to be described associating hypoplastic olfactory bulbs with pituitary stalk interruption. This has challenged both concepts, suggesting that sometimes SK and pituitary stalk interruption syndrome may share a common genetic origin and pathophysiology.